
Le notizie in evidenza
Approvazione della Commissione Europea del farmaco MITAPIVAT (Pyrukynd) di Agios Pharmaceuticals

CAMBRIDGE, Massachusetts, 22 maggio 2026 𝗔𝗴𝗶𝗼𝘀 𝗣𝗵𝗮𝗿𝗺𝗮𝗰𝗲𝘂𝘁𝗶𝗰𝗮𝗹𝘀, 𝗮𝘇𝗶𝗲𝗻𝗱𝗮 𝗯𝗶𝗼𝗳𝗮𝗿𝗺𝗮𝗰𝗲𝘂𝘁𝗶𝗰𝗮 𝗳𝗼𝗰𝗮𝗹𝗶𝘇𝘇𝗮𝘁𝗮 𝘀𝘂𝗹𝗹𝗮 𝗳𝗼𝗿𝗻𝗶𝘁𝘂𝗿𝗮 𝗱𝗶 𝗳𝗮𝗿𝗺𝗮𝗰𝗶 𝗶𝗻𝗻𝗼𝘃𝗮𝘁𝗶𝘃𝗶 𝗽𝗲𝗿 𝗽𝗮𝘇𝗶𝗲𝗻𝘁𝗶 𝗮𝗳𝗳𝗲𝘁𝘁𝗶 𝗱𝗮 𝗺𝗮𝗹𝗮𝘁𝘁𝗶𝗲 𝗿𝗮𝗿𝗲, 𝗵𝗮 𝗮𝗻𝗻𝘂𝗻𝗰𝗶𝗮𝘁𝗼 𝗼𝗴𝗴𝗶 𝗰𝗵𝗲 𝗹𝗮 𝗖𝗼𝗺𝗺𝗶𝘀𝘀𝗶𝗼𝗻𝗲 𝗲𝘂𝗿𝗼𝗽𝗲𝗮 (𝗖𝗘) 𝗵𝗮 𝗰𝗼𝗻𝗰𝗲𝘀𝘀𝗼 𝗹'𝗮𝘂𝘁𝗼𝗿𝗶𝘇𝘇𝗮𝘇𝗶𝗼𝗻𝗲 𝗮𝗹𝗹'𝗶𝗺𝗺𝗶𝘀𝘀𝗶𝗼𝗻𝗲 𝗶𝗻 𝗰𝗼𝗺𝗺𝗲𝗿𝗰𝗶𝗼 𝗱𝗶 𝗣𝗬𝗥𝗨𝗞𝗬𝗡𝗗® (𝗠𝗶𝘁𝗮𝗽𝗶𝘃𝗮𝘁), 𝘂𝗻 𝗮𝘁𝘁𝗶𝘃𝗮𝘁𝗼𝗿𝗲 𝗼𝗿𝗮𝗹𝗲 𝗱𝗲𝗹𝗹𝗮 𝗽𝗶𝗿𝘂𝘃𝗮𝘁𝗼 𝗰𝗵𝗶𝗻𝗮𝘀𝗶 (𝗣𝗞), 𝗻𝗲𝗴𝗹𝗶 𝗮𝗱𝘂𝗹𝘁𝗶 𝗽𝗲𝗿 𝗶𝗹 𝘁𝗿𝗮𝘁𝘁𝗮𝗺𝗲𝗻𝘁𝗼 𝗱𝗲𝗹𝗹'𝗮𝗻𝗲𝗺𝗶𝗮 𝗮𝘀𝘀𝗼𝗰𝗶𝗮𝘁𝗮 𝗮𝗱 𝗮𝗹𝗳𝗮 𝗼 𝗯𝗲𝘁𝗮-𝘁𝗮𝗹𝗮𝘀𝘀𝗲𝗺𝗶𝗮 𝘁𝗿𝗮𝘀𝗳𝘂𝘀𝗶𝗼𝗻𝗲-𝗱𝗶𝗽𝗲𝗻𝗱𝗲𝗻𝘁𝗲 𝗲 𝗻𝗼𝗻 𝘁𝗿𝗮𝘀𝗳𝘂𝘀𝗶𝗼𝗻𝗲 𝗱𝗶𝗽𝗲𝗻𝗱𝗲𝗻𝘁𝗲, 𝗰𝗼𝗻 𝗱𝗲𝘀𝗶𝗴𝗻𝗮𝘇𝗶𝗼𝗻𝗲 𝗱𝗶 𝗺𝗲𝗱𝗶𝗰𝗶𝗻𝗮𝗹𝗲 𝗼𝗿𝗳𝗮𝗻𝗼.
La talassemia impone un profondo fardello quotidiano ai pazienti, aggravato dalla mancanza di opzioni terapeutiche accessibili per tutte le forme della malattia”, ha affermato Maria Domenica Cappellini, MD, Professoressa di Medicina Interna, Università degli Studi di Milano, Italia e ricercatore nel programma clinico di Fase 3 di PYRUKYND per la talassemia. "I risultati della Fase 3 dimostrano il potenziale di PYRUKYND nell'affrontare questo problema e migliorare gli esiti per coloro che ne hanno bisogno, indipendentemente dal loro genotipo o stato trasfusionale. Questa approvazione rappresenta un importante passo avanti che può aiutare migliaia di pazienti in tutta l'UE a gestire questa malattia debilitante e potenzialmente letale.
La decisione della Commissione europea si basa sui risultati degli studi globali di fase 3 ENERGIZE e ENERGIZE-T, randomizzati, in doppio cieco e controllati con placebo, condotti su adulti affetti rispettivamente da alfa o beta talassemia non trasfusione dipendente e trasfusione-dipendente.
L'approvazione di PYRUKYND da parte della Commissione Europea è un traguardo importante, che segna un progresso significativo per i pazienti affetti da talassemia", ha affermato Brian Goff, Amministratore delegato, Agios. Non vediamo l'ora di proseguire la nostra collaborazione con Avanzanite per rendere PYRUKYND disponibile ai pazienti affetti da talassemia in tutta l'UE, rafforzando il nostro impegno a promuovere l'innovazione per la comunità globale."
Nel 2025, Agios ha stipulato un accordo esclusivo con Avanzanite Bioscience BV (Avanzanite) per la commercializzazione e la distribuzione di PYRUKYND in tutto lo Spazio economico europeo, Regno Unito, Svizzera. Sede centrale ad Amsterdam, Avanzanite è un'azienda farmaceutica specializzata in fase commerciale dedicata a portare farmaci per malattie rare ai pazienti in tutta Europa.
𝗠𝗶𝘁𝗮𝗽𝗶𝘃𝗮𝘁 è ora approvato per gli adulti affetti da talassemia nell'UE, Arabia Saudita, Emirati Arabi Uniti, sotto il marchio PYRUKYND, nonché negli Stati Uniti con il nome commerciale AQVESME™ (mitapivat).
Mitapivat è un attivatore della piruvato chinasi che migliora il metabolismo energetico dei globuli rossi. Aumentando la produzione di energia intracellulare, consente ai globuli rossi di funzionare in modo più efficiente e di sopravvivere più a lungo, determinando livelli di emoglobina più elevati. A differenza delle trasfusioni, che forniscono una sostituzione temporanea dei globuli rossi, mitapivat offre un approccio orale giornaliero, basato sul meccanismo d'azione , che mira a correggere l'inefficienza metabolica alla base dell'anemia. Questo può potenzialmente ridurre l'affaticamento e, in alcuni pazienti, diminuire il fabbisogno trasfusionale. Gli studi hanno dimostrato miglioramenti nei livelli di emoglobina, riduzioni clinicamente significative del fabbisogno trasfusionale e migliori risultati in termini di affaticamento rispetto al placebo. Questi risultati posizionano il mitapivat come una nuova importante opzione nella gestione della talassemia negli adulti. (ndr).
Dopo l'autorizzazione all'immissione in commercio rilasciata dalla Commissione Europea, il farmaco entra in una fase distinta: la valutazione nazionale. In Italia è l'Agenzia Italiana del Farmaco a stabilire se e a quali condizioni il medicinale sarà rimborsato dal Servizio Sanitario Nazionale. Ogni Stato membro deve completare i processi nazionali di valutazione dell'HTA (Health Technology Assessment), negoziazione del prezzo e rimborsabilità per consentirne l'accesso tramite il sistema sanitario pubblico. Le autorità sanitarie nazionali valutano il valore aggiunto clinico del farmaco per stabilirne il prezzo di vendita e decidere se (e in quale percentuale) i costi debbano essere coperti dal sistema pubblico. In Italia, l'approvazione dell'EMA avvia la fase di competenza dell'AIFA (Agenzia Italiana del Farmaco). L'iter prevede: Negoziazione: AIFA e l'azienda farmaceutica negoziano il prezzo finale e i criteri di rimborsabilità a carico del Servizio Sanitario Nazionale (SSN). Classificazione: AIFA decide la classe di rimborsabilità (es. Classe H per uso ospedaliero o Classe A per farmaci dispensabili in farmacia). Determinazione dell'Innovatività: L'ente valuta se il farmaco possiede i requisiti per essere riconosciuto come "farmaco innovativo", accedendo a un fondo dedicato per garantirne la somministrazione immediata.
CHMP: parere positivo per PYRUKYNDⓇ (Mitapivat) per adulti con talassemia
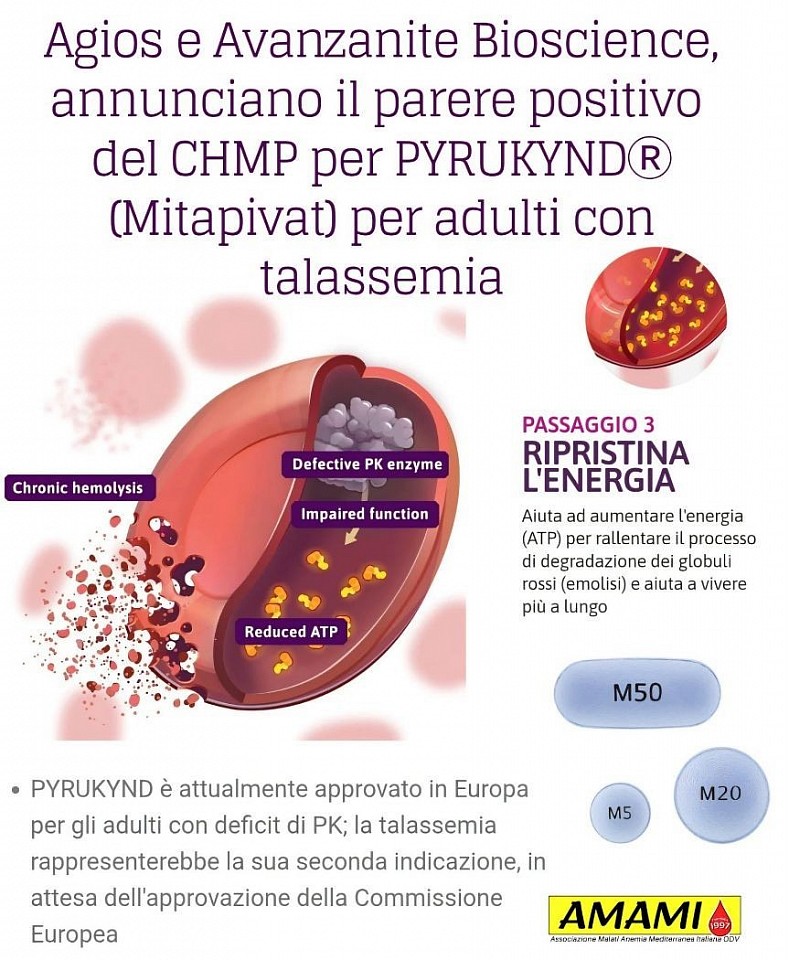
𝗔𝗴𝗶𝗼𝘀 𝗣𝗵𝗮𝗿𝗺𝗮𝗰𝗲𝘂𝘁𝗶𝗰𝗮𝗹𝘀 𝗲 𝗶𝗹 𝘀𝘂𝗼 𝗽𝗮𝗿𝘁𝗻𝗲𝗿 𝗔𝘃𝗮𝗻𝘇𝗮𝗻𝗶𝘁𝗲 𝗕𝗶𝗼𝘀𝗰𝗶𝗲𝗻𝗰𝗲 𝗕𝗩, 𝗮𝘇𝗶𝗲𝗻𝗱𝗮 𝗳𝗮𝗿𝗺𝗮𝗰𝗲𝘂𝘁𝗶𝗰𝗮 𝗲𝘂𝗿𝗼𝗽𝗲𝗮, 𝗵𝗮𝗻𝗻𝗼 𝗶𝗻𝗳𝗼𝗿𝗺𝗮𝘁𝗼 𝗰𝗵𝗲 𝗶𝗹 𝗖𝗼𝗺𝗶𝘁𝗮𝘁𝗼 𝗽𝗲𝗿 𝗶 𝗺𝗲𝗱𝗶𝗰𝗶𝗻𝗮𝗹𝗶 𝗽𝗲𝗿 𝘂𝘀𝗼 𝘂𝗺𝗮𝗻𝗼 (𝗖𝗛𝗠𝗣) 𝗱𝗲𝗹𝗹'𝗔𝗴𝗲𝗻𝘇𝗶𝗮 𝗲𝘂𝗿𝗼𝗽𝗲𝗮 𝗽𝗲𝗿 𝗶 𝗺𝗲𝗱𝗶𝗰𝗶𝗻𝗮𝗹𝗶 (𝗘𝗠𝗔) 𝗵𝗮 𝗮𝗱𝗼𝘁𝘁𝗮𝘁𝗼 𝘂𝗻 𝗽𝗮𝗿𝗲𝗿𝗲 𝗽𝗼𝘀𝗶𝘁𝗶𝘃𝗼 𝗽𝗲𝗿 𝗹𝗮 𝗻𝘂𝗼𝘃𝗮 𝗶𝗻𝗱𝗶𝗰𝗮𝘇𝗶𝗼𝗻𝗲 𝗱𝗶 𝗠𝗶𝘁𝗮𝗽𝗶𝘃𝗮𝘁, 𝘂𝗻 𝗮𝘁𝘁𝗶𝘃𝗮𝘁𝗼𝗿𝗲 𝗼𝗿𝗮𝗹𝗲 𝗱𝗲𝗹𝗹𝗮 𝗽𝗶𝗿𝘂𝘃𝗮𝘁𝗼 𝗰𝗵𝗶𝗻𝗮𝘀𝗶 (𝗣𝗞), 𝗻𝗲𝗴𝗹𝗶 𝗮𝗱𝘂𝗹𝘁𝗶 𝗽𝗲𝗿 𝗶𝗹 𝘁𝗿𝗮𝘁𝘁𝗮𝗺𝗲𝗻𝘁𝗼 𝗱𝗲𝗹𝗹'𝗮𝗻𝗲𝗺𝗶𝗮 𝗮𝘀𝘀𝗼𝗰𝗶𝗮𝘁𝗮 𝗮 𝘁𝗿𝗮𝘀𝗳𝘂𝘀𝗶𝗼𝗻𝗲 𝗱𝗶𝗽𝗲𝗻𝗱𝗲𝗻𝘁𝗲 𝗲 𝗮𝗹𝗳𝗮 𝗼 𝗯𝗲𝘁𝗮-𝘁𝗮𝗹𝗮𝘀𝘀𝗲𝗺𝗶𝗮 𝗻𝗼𝗻 𝘁𝗿𝗮𝘀𝗳𝘂𝘀𝗶𝗼𝗻𝗲-𝗱𝗶𝗽𝗲𝗻𝗱𝗲𝗻𝘁𝗲. 𝗟𝗮 𝘁𝗲𝗿𝗮𝗽𝗶𝗮 𝗲' 𝗮𝘁𝘁𝘂𝗮𝗹𝗺𝗲𝗻𝘁𝗲 𝗮𝗽𝗽𝗿𝗼𝘃𝗮𝘁𝗮 𝗶𝗻 𝗘𝘂𝗿𝗼𝗽𝗮 𝗽𝗲𝗿 𝗴𝗹𝗶 𝗮𝗱𝘂𝗹𝘁𝗶 𝗰𝗼𝗻 𝗱𝗲𝗳𝗶𝗰𝗶𝘁 𝗱𝗶 𝗣𝗞; 𝗹𝗮 𝘁𝗮𝗹𝗮𝘀𝘀𝗲𝗺𝗶𝗮 𝗿𝗮𝗽𝗽𝗿𝗲𝘀𝗲𝗻𝘁𝗲𝗿𝗲𝗯𝗯𝗲 𝗹𝗮 𝘀𝘂𝗮 𝘀𝗲𝗰𝗼𝗻𝗱𝗮 𝗶𝗻𝗱𝗶𝗰𝗮𝘇𝗶𝗼𝗻𝗲, 𝗶𝗻 𝗮𝘁𝘁𝗲𝘀𝗮 𝗱𝗲𝗹𝗹'𝗮𝗽𝗽𝗿𝗼𝘃𝗮𝘇𝗶𝗼𝗻𝗲 𝗱𝗲𝗹𝗹𝗮 𝗖𝗼𝗺𝗺𝗶𝘀𝘀𝗶𝗼𝗻𝗲 𝗘𝘂𝗿𝗼𝗽𝗲𝗮
✅️"Il parere positivo del CHMP per Mitapivat rappresenta un passo fondamentale verso la disponibilità di questo trattamento per i pazienti adulti affetti da talassemia, una comunità che ne ha davvero bisogno. – Ha dichiarato il Dr. Mark Bechter, Senior Vice President of Medical Affairs di Avanzanite – Non vediamo l'ora di lavorare con Agios per garantire l'accesso a questa terapia ai pazienti di tutta Europa, in attesa dell'approvazione da parte della Commissione Europea". La Commissione europea esaminerà ora il parere del CHMP e Agios ha indicato che la decisione finale è prevista per l'inizio del 2026. Il parere positivo del CHMP si basa sui risultati degli studi globali, randomizzati, in doppio cieco, controllati con placebo ENERGIZE-T e ENERGIZE di Fase 3 in adulti con alfa o beta-talassemia trasfusione dipendente e non trasfusione dipendente, rispettivamente.
✅️Lo studio ENERGIZE-T ha randomizzato 258 pazienti dipendenti da trasfusioni 2:1 a ricevere mitapivat 100 mg due volte al giorno o placebo. L'endpoint primario era la percentuale di pazienti che ottenevano una risposta di riduzione trasfusionale (TRR), definita come una riduzione del ≥50% dei pazienti trasfusi unità di globuli rossi (RBC) con una riduzione di ≥2 unità di globuli rossi trasfusi in un periodo continuo di 12 settimane fino alla settimana 48. Diverse misure aggiuntive di riduzione delle trasfusioni sono state incluse come endpoint secondari chiave e il raggiungimento dell'indipendenza trasfusionale è stato un endpoint secondario. Lo studio ha anche valutato la sicurezza e la tollerabilità.
✅️Lo studio ENERGIZE ha randomizzato 194 pazienti non dipendenti dalle trasfusioni 2:1 a ricevere mitapivat 100 mg due volte al giorno o placebo. L'endpoint primario era la percentuale di pazienti che ottenevano una risposta all'emoglobina, definita come un aumento di ≥1,0 g/dL delle concentrazioni medie di emoglobina dalla settimana 12 alla settimana 24 rispetto al basale. Gli endpoint secondari chiave includevano le variazioni rispetto al basale nella valutazione funzionale delle malattie croniche Punteggi di Therapy-Fatigue (FACIT-Fatigue) e nella concentrazione media di emoglobina dalla settimana 12 alla settimana 24. Lo studio ha anche valutato la sicurezza e la tollerabilità. Nel giugno 2025, Avanzanite ha stipulato un accordo esclusivo con Agios per la commercializzazione e la distribuzione della terapia nello Spazio Economico Europeo, nel Regno Unito e in Svizzera.
Fonte https://avanzanite.com/index
Editing genomico: finalmente l’AIFA dà il via libera alla rimborsabilità di CASGEVY

Il 17 settembre 2025 segna una data storica per la medicina italiana e per i pazienti affetti da gravi malattie ereditarie del sangue. In quella giornata, infatti, il Consiglio di Amministrazione dell’Agenzia Italiana del Farmaco (AIFA) ha approvato la rimborsabilità di Casgevy (exagamglogene autotemcel), la prima terapia genica basata su CRISPR autorizzata in Europa e in Italia. Si tratta di un traguardo che unisce la ricerca scientifica più avanzata con la concretezza dell’accesso terapeutico: da oggi, i pazienti italiani con beta-talassemia trasfusione-dipendente (TDT) e anemia falciforme (SCD) potranno ricevere un trattamento capace di modificare radicalmente il decorso della loro malattia, senza doversi più affidare unicamente a trasfusioni o terapie di supporto.
UNA NUOVA FRONTIERA DELLA MEDICINA
Le due patologie per cui Casgevy è indicato hanno un’origine comune: mutazioni del gene della beta-globina, che compromettono la corretta produzione di emoglobina. Nella beta-talassemia, questo difetto rende necessarie trasfusioni regolari e continue per tutta la vita; nell’anemia falciforme, la malformazione dei globuli rossi causa crisi vaso-occlusive dolorose e potenzialmente letali.
Exagamglogene autotemcel introduce un approccio radicalmente diverso rispetto alle terapie tradizionali. Le cellule staminali ematopoietiche del paziente vengono prelevate e successivamente modificate in laboratorio attraverso la tecnica CRISPR, che consente di “correggere” con estrema precisione le sequenze difettose del DNA. Una volta reinfuse, queste cellule ripopolano il midollo osseo e danno origine a una nuova produzione di cellule del sangue in grado di sintetizzare emoglobina fetale, una forma di emoglobina sana e funzionale. Il risultato è la riduzione o, in molti casi, l’eliminazione della necessità di trasfusioni per la talassemia e la drastica diminuzione delle crisi vaso-ostruttive nell’anemia falciforme.
I PRIMI PAZIENTI TRATTATI IN ITALIA
L’Italia non è rimasta a guardare in attesa dell’approvazione formale della rimborsabilità. Già durante la scorsa primavera, quattro pazienti sono stati sottoposti a trattamento con Casgevy in centri altamente specializzati a Perugia, Pavia e Pescara, grazie al percorso di accesso precoce di AIFA. Tre di loro erano affetti da anemia falciforme e uno da talassemia. L’esito è stato incoraggiante: i pazienti hanno mostrato benefici clinici significativi e hanno potuto riprendere progressivamente le loro attività quotidiane. Una testimonianza concreta di come le terapie avanzate possano tradursi in un miglioramento reale e tangibile della qualità di vita.
INNOVATIVITÀ RICONOSCIUTA E MONITORAGGIO COSTANTE
Casgevy è stato anche riconosciuto come farmaco innovativo, un’etichetta che non solo sottolinea il valore terapeutico e scientifico della terapia, ma consente anche un accesso prioritario e agevolato per i pazienti. L’inserimento in un registro di monitoraggio garantirà inoltre che ogni trattamento venga seguito con attenzione, raccogliendo dati su efficacia e sicurezza nel tempo, a tutela sia dei pazienti sia della sostenibilità del Servizio Sanitario Nazionale.
LA QUESTIONE DEL COSTO E LA PROSPETTIVA DI LUNGO PERIODO
Non si può ignorare l’aspetto economico: Casgevy ha un costo stimato di circa due milioni di euro per paziente. Una cifra che, a prima vista, appare enorme. Tuttavia, bisogna considerare che si tratta di una terapia somministrata una sola volta, con effetti potenzialmente duraturi, capace di sostituire anni di cure croniche, trasfusioni frequenti, ricoveri e complicanze. In quest’ottica, come ha sottolineato lo stesso Ministro della Salute, il costo non è solo una spesa ma un investimento: per il sistema sanitario, che potrebbe risparmiare nel lungo termine, e per la società, che guadagna cittadini più sani e autonomi.
UNA RIVOLUZIONE CHE PARTE DALL’ITALIA
Con l’approvazione di Casgevy, l’Italia si pone all’avanguardia nel campo delle terapie avanzate. È una rivoluzione che non riguarda soltanto la talassemia e l’anemia falciforme, ma che apre la strada a un’intera generazione di trattamenti basati sull’editing genetico. Per i pazienti e le loro famiglie, questa approvazione rappresenta molto più che una notizia di cronaca sanitaria: significa poter guardare al futuro con una speranza nuova, concreta e finalmente accessibile grazie al Servizio sanitario nazionale.
Vertex presenta nuovi dati positivi a lungo termine per terapia genica
16 giugno 2025 - Vertex Pharmaceuticals Incorporated (Nasdaq: VRTX) ha annunciato i nuovi dati positivi a lungo termine relativi a exagamglogene autotemcel (exa-cel) derivanti da studi clinici globali condotti su persone affette da anemia falciforme severa (SCD) o beta-talassemia trasfusione-dipendente (TDT). I risultati, presentati in occasione del Congresso annuale dell’Associazione Europea di Ematologia (EHA), svoltosi dal 13 - 16 giugno a Milano, continuano a dimostrare i benefici clinici significativi e duraturi di exa-cel. Il follow-up più lungo nei pazienti con SCD ha superato ormai i 5.5 anni, che diventano oltre 6 nei pazienti con TDT, con una media rispettivamente di 39.4 e 43.5 mesi. Exa-cel è la prima e unica terapia approvata di editing genetico basata sulla tecnologia CRISPR/Cas9.
“Questi dati a lungo termine confermano ulteriormente che exa-cel può offrire significativi benefici clinici, duraturi nel tempo, alle persone eleggibili al trattamento affette da anemia falciforme o beta-talassemia trasfusione-dipendente”, ha affermato Franco Locatelli, M.D., Ph.D., Professore di Pediatria presso l’Università Cattolica del Sacro Cuore di Roma, Direttore del Dipartimento di Ematologia e Oncologia Pediatrica dell’Ospedale Pediatrico Bambino Gesù, Presidente del Comitato Direttivo del Programma TDT di Vertex e autore principale dei dati clinici su exa-cel nella TDT presentati al Congresso EHA.
I nuovi dati di follow-up a lungo termine provenienti dagli studi clinici su exa-cel presentati al Congresso:
SCD: 43/45 (95.6%) dei pazienti valutabili con almeno 16 mesi di follow-up sono risultati liberi da crisi vaso-occlusive (VOC) per almeno 12 mesi consecutivi (VF12) negli studi CLIMB-121 e CLIMB-131 combinati (IC 95%: 84,9%, 99,5%). La durata media del periodo senza VOC è stata di 35.0 mesi (intervallo: 14.4 – 66.2 mesi).
Tutti i pazienti valutabili (45/45 [100%]) hanno ottenuto l’assenza di ricoveri ospedalieri per VOC gravi per almeno 12 mesi consecutivi (HF12) negli studi CLIMB-121 e CLIMB-131 combinati (IC 95%: 92,1%, 100%), con una media senza ospedalizzazione di 36.1 mesi (intervallo: 14.5 – 66.2 mesi).
TDT: 54/55 (98.2%) dei pazienti valutabili con almeno 16 mesi di follow-up hanno ottenuto l’indipendenza da trasfusioni per almeno 12 mesi consecutivi, con un valore medio ponderato di emoglobina (Hb) di almeno 9 g/dl (TI12) negli studi CLIMB-111 e CLIMB-131 combinati (IC 95%: 90,3%, 100%). La durata media dell’indipendenza da trasfusioni è stata di 40,5 mesi (intervallo: 13.6 – 70.8 mesi).
L’unico paziente valutabile che non ha ottenuto TI12 è rimasto libero da trasfusioni per 14.8 mesi.
La terapia di chelazione del ferro è stata sospesa per più di 6 mesi in 39/56 (69.6%) dei pazienti trattati in seguito all’infusione di exa-cel, con un sostenuto miglioramento dei livelli di ferritina e di ferro epatico, il che suggerisce che exa-cel abbia il potenziale per correggere un’eritropoiesi inefficace. I pazienti continuano a presentare livelli stabili di emoglobina fetale (HbF) e di editing allelico. Il profilo di sicurezza di exa-cel continua a essere generalmente coerente con il condizionamento mieloablativo a base di busulfano e con il trapianto autologo di cellule staminali emopoietiche.
Grazie agli accordi di rimborsabilità, Vertex ha ottenuto l’accesso al trattamento per i pazienti eleggibili per SCD o TDT in diversi paesi, tra cui Austria, Bahrein, Danimarca, Inghilterra, Arabia Saudita, Irlanda del Nord, Scozia, Emirati Arabi Uniti, Stati Uniti e Galles. Vertex continua a collaborare a livello globale con gli enti governativi e le autorità preposte alla rimborsabilità, per garantire un accesso sostenibile ad altri pazienti eleggibili.
Casgevy® è una terapia cellulare non virale, costituita da cellule modificate ex vivo mediante la tecnologia CRISPR/Cas9, per pazienti eleggibili con SCD o TDT, in cui le cellule staminali e progenitrici ematopoietiche del paziente stesso vengono modificate nella regione enhancer specifica per gli eritroidi del gene BCL11A attraverso una precisa rottura del doppio filamento del DNA. Questa modifica porta alla produzione di alti livelli di emoglobina fetale (HbF; emoglobina F) nei globuli rossi. L’HbF è la forma di emoglobina che trasporta ossigeno, naturalmente presente durante lo sviluppo fetale, che viene sostituita dalla forma adulta dell’emoglobina dopo la nascita.
CASGEVY, la terapia genica per la B-talassemia e l'anemia falciforme
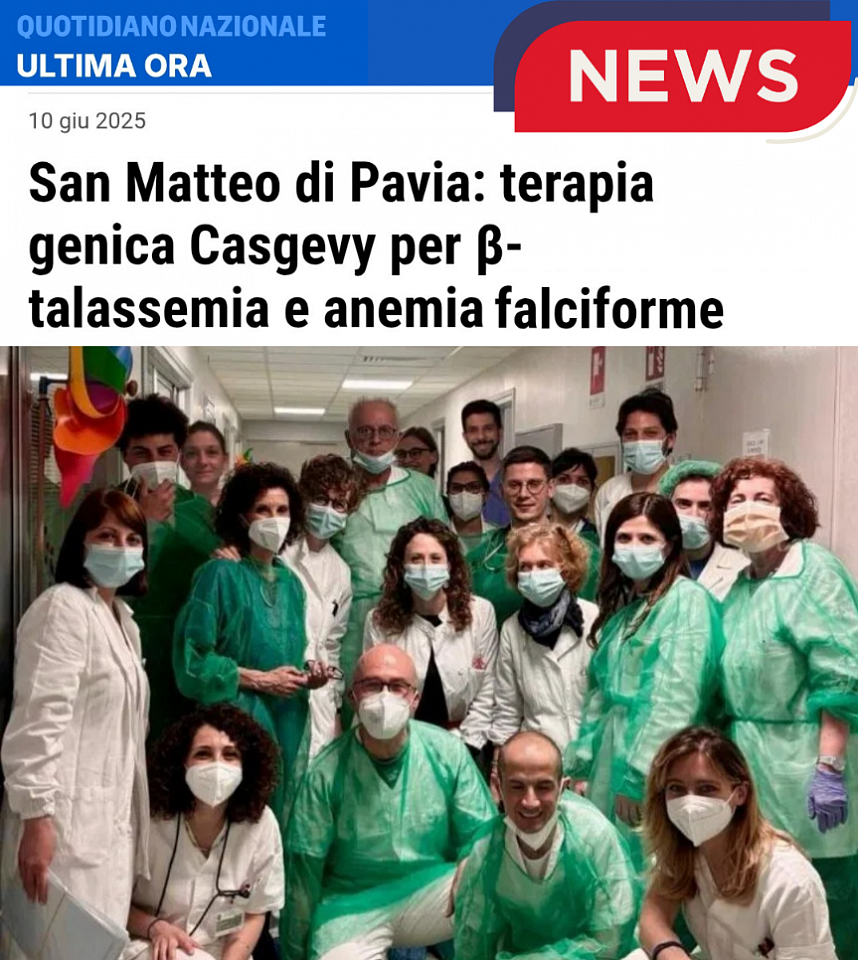
PAVIA – Al Policlinico San Matteo di Pavia è stata scritta una nuova pagina della medicina nella cura delle emoglobinopatie. Due giovani pazienti ventenni, affette rispettivamente da ß-talassemia major e anemia falciforme grave, sono state le prime in Italia e Europa a ricevere Casgevy, la terapia genica basata sulla tecnologia CRISPR-Cas9, al di fuori delle sperimentazioni cliniche.
Martedì 13 maggio 2025 la paziente con ß-talassemia è stata la prima in Europa a ricevere l’infusione di Casgevy, al di fuori di sperimentazioni cliniche. La ragazza sta bene, anche se è ancora ricoverata in attesa di poter essere dimessa nelle prossime settimane. Giovedì 22 maggio 2025 la paziente con anemia falciforme è stata la prima in Italia a ricevere l’infusione di Casgevy, al di fuori di sperimentazioni cliniche. Dopo un ricovero di oltre un mese presso la Sezione Trapianto di Midollo Osseo dell’Oncoematologia Pediatrica, la ragazza è stata dimessa il 3 giugno, ed ora prosegue i controlli in ambulatorio.
Il trattamento innovativo rappresenta una svolta nella cura delle emoglobinopatie, malattie genetiche gravi e invalidanti. La ß-talassemia e l’anemia falciforme sono gravi emoglobinopatie ereditarie, causate da mutazioni del gene della ß-globina. Casgevy agisce attraverso un approccio innovativo di editing genetico ex vivo delle cellule staminali ematopoietiche autologhe, volto a riattivare la produzione di emoglobina fetale (HbF), capace di compensare efficacemente il difetto genetico.
Il processo terapeutico prevede la raccolta delle cellule staminali del paziente tramite aferesi, il loro editing genetico effettuato dall’azienda farmaceutica, una fase di chemioterapia mieloablativa e infine la reinfusione delle cellule corrette.
Nel maggio 2024 è stata inoltrata ad AIFA la richiesta di accesso al Fondo Nazionale AIFA 5% per il trattamento delle due pazienti, in assenza di donatori idonei e in considerazione del rapido peggioramento clinico. Le richieste sono state approvate, consentendo di procedere con la raccolta delle cellule staminali nel corso della seconda metà del 2024.
Fonte https://www.quotidiano.net/
MITAPIVAT: nuovo farmaco orale per la Talassemia

Un nuovo farmaco orale potrebbe presto entrare a far parte delle opzioni terapeutiche per chi convive con la talassemia. Si chiama mitapivat e ha già dimostrato la sua efficacia in un'altra malattia del sangue, il deficit di piruvato chinasi. Dai risultati degli studi clinici più recenti, è emerso che questo trattamento porta benefici anche ai pazienti talassemici, sia a quelli che dipendono dalle trasfusioni sia a chi non ne ha bisogno.
La dottoressa Raffaella Origa, presidente della SITE e professoressa associata di Pediatria all'Università di Cagliari, ci spiega meglio come funziona questo nuovo farmaco: Mitapivat è una piccola molecola che attiva la piruvato chinasi, un enzima fondamentale nella glicolisi, cioè la via principale con cui i globuli rossi producono energia sotto forma di ATP (adenosina trifosfato). Quando questo enzima è carente, come nel deficit di piruvato chinasi (una malattia ereditaria causata da mutazioni del gene PKLR), i globuli rossi si distruggono prima del tempo, causando un’anemia emolitica cronica di diversa gravità. Mitapivat, attivando l’enzima alterato, è in grado di aumentare i livelli di emoglobina nei pazienti con deficit non trasfusione-dipendente e di ridurre la necessità di trasfusioni in quelli con la forma trasfusione-dipendente. Poiché mitapivat agisce anche sull’enzima normale (wild-type), è stato testato anche in altre forme di anemia, tra cui la talassemia. In questa condizione, dove i globuli rossi hanno un fabbisogno energetico aumentato e quindi un’attività piruvato-chinasica relativamente bassa, si è ipotizzato e poi confermato che l’aumento di ATP prodotto dall’attivazione dell’enzima può migliorare la resistenza dei globuli rossi allo stress ossidativo, aiutandoli a sopravvivere più a lungo. Inoltre, oltre ai benefici sul metabolismo energetico, mitapivat sembra ridurre il danno ossidativo nei precursori dei globuli rossi, favorendo una maturazione migliore e una maggiore sopravvivenza degli eritroblasti, con un conseguente miglioramento dell’efficacia dell’eritropoiesi.
Dopo uno studio di Fase II che aveva mostrato come mitapivat aumenti di almeno 1 g/dL i livelli di emoglobina in pazienti adulti con talassemia non trasfusione-dipendente, sono stati avviati gli studi Energize e Energize T per approfondire efficacia e tollerabilità del farmaco. Come molti studi di Fase III, sono stati condotti in più centri, con un numero maggiore di pazienti, e sono randomizzati e controllati, confrontando mitapivat con un placebo. Una particolarità di questi studi è che hanno coinvolto sia pazienti con beta talassemia sia con alfa talassemia.
Entrambi gli studi hanno dato risultati molto positivi, raggiungendo gli obiettivi principali e secondari. In dettaglio, tra 55 pazienti con talassemia non trasfusione-dipendente trattati con mitapivat, il 42,3% ha registrato un aumento di emoglobina di almeno 1 g/dL tra la settimana 12 e 24 (con una media di +1,56 g/dL), mentre solo 1 paziente su 64 nel gruppo placebo ha avuto questo risultato. La risposta è stata superiore al placebo in tutti i sottogruppi. Inoltre, i marker di emolisi sono diminuiti e il trattamento ha migliorato sia il benessere percepito dai pazienti sia la loro capacità funzionale, misurata con il test del cammino in 6 minuti. Tra i pazienti con talassemia trasfusione-dipendente, il 30,4% di quelli trattati con mitapivat ha ridotto di almeno il 50% le trasfusioni necessarie, contro il 12,6% del gruppo placebo. La differenza è stata quindi molto significativa. In più, 17 pazienti trattati con mitapivat sono diventati trasfusione-indipendenti, cioè hanno avuto almeno 8 settimane consecutive senza trasfusioni. Il farmaco è stato ben tollerato, con effetti collaterali più comuni come cefalea, insonnia e nausea, presenti in entrambi gli studi. Se questi dati verranno confermati nella pratica clinica, mitapivat potrebbe diventare un’opzione terapeutica importante per i pazienti con talassemia, sia trasfusione-dipendente che no. Nella forma non trasfusione-dipendente, l’aumento dell’emoglobina migliora la qualità della vita e riduce il rischio di complicanze. Anche nella talassemia trasfusione-dipendente, dove il grado di anemia è un fattore prognostico importante, mitapivat potrebbe portare benefici. Ridurre le trasfusioni non solo rende la vita quotidiana più semplice, ma può anche limitare l’accumulo di ferro (con dati preliminari promettenti), le sue complicanze e la necessità di terapie ferrochelanti, oltre a diminuire il carico sul sistema sangue. La somministrazione orale giornaliera, che si può fare comodamente a casa, è un ulteriore vantaggio in termini di praticità. Poiché gli effetti collaterali finora sembrano gestibili, mitapivat potrebbe essere indicato anche per pazienti non candidabili ad altre terapie recentemente approvate. Infine, va ricordato che il farmaco ha mostrato efficacia e sicurezza anche nell’alfa-talassemia, una forma finora priva di trattamenti specifici in grado di migliorare concretamente la qualità della vita.
Fonte www.osservatoriomalattierare.it
La terapia genica per la Talassemia
Quando sarà disponibile anche in Italia la terapia genica per la cura della β-talassemia e dell’anemia falciforme? La prima terapia di editing genetico sviluppata con la tecnologia Crispr/Cas 9, nonché l’unica disponibile in Europa, a differenza degli Stati Uniti, dove sul mercato ci sono anche betibeglogene autotemcel e lovo-cel (entrambe di Bluebird Bio), è attualmente oggetto di negoziazione tra l’azienda che ha sviluppato la sofisticata terapia genica (Vertex Pharmaceuticals) e l’Agenzia italiana del farmaco (Aifa) da quasi un anno. Nell’arco di 12 mesi presumibilmente sono attesi sia l’ok alla rimborsabilità del trattamento, attualmente in Europa disponibile soltanto in Austria e Danimarca, al di là del Regno Unito, sia l’avvio delle prime somministrazioni.
Exa-cel, questo il nome della terapia genica sviluppata da Vertex assieme a Crispr Therapeutics, si è confermata efficace nel tempo nel mantenimento dei due obiettivi primari dello studio. Ovvero: la possibilità di non dover più sottoporre i pazienti talassemici che ne hanno bisogno alle periodiche trasfusioni di sangue (all’incirca il 70 per cento dei settemila talassemici italiani) e l’assenza di crisi vaso-occlusive nei pazienti con anemia falciforme. Una volta raggiunta l’intesa economica e definito il range d’età dei pazienti trattabili – per l’Ema nessun problema dai 12 anni in su, ma i soggetti arruolati nello studio pivotal non avevano più di 35 anni – occorrerà definire la mappa dei centri autorizzati alla somministrazione della terapia. Almeno inizialmente, la procedura sarà infatti centralizzata in un numero definito di ospedali, che dovranno rispondere ai requisiti che saranno fissati dall’Aifa.
Come primo passo, ogni persona candidata al trattamento (“one shot”, con una somministrazione potenzialmente efficace per tutta la vita) sarà sottoposta a un paio di procedure di aferesi per prelevare le cellule staminali ematopoietiche da sottoporre a editing genetico in laboratorio. I tempi di questa procedura non saranno brevi: da quattro a sei mesi. A seguire, con le cellule modificate pronte per essere reinfuse, “i pazienti saranno sottoposti a una chemioterapia a base di busulfano per cinque giorni, per distruggere il vecchio midollo talassemico e preparare l’organismo a ricevere quello geneticamente modificato per iniziare a produrre l’emoglobina sana”.
fonte https://www.aboutpharma.com/sanita
Nuovi dati a lungo termine di Casgevy, la terapia di editing genetico basata su CRISPR/Cas9.
Vertex Pharmaceuticals Incorporated ha annunciato i dati a lungo termine di Casgevy® (exagamglogene autotemcel, exa-cel) provenienti dagli studi clinici globali condotti su persone affette da anemia falciforme SCD o beta-talassemia trasfusione-dipendente (TDT). Exa-cel è la prima e unica terapia di editing genetico basata su CRISPR/Cas9.
I risultati, presentati all'Annuale Meeting dell’American Society of Hematology ASH 2024, continuano a dimostrare i benefici clinici duraturi e trasformativi di exa-cel. Il follow-up più esteso per i pazienti con SCD e TDT è ora superiore ai 5 anni, con una mediana rispettivamente di 33,2 mesi e 38,1 mesi. “Questi dati forniscono un’ulteriore dimostrazione dei benefici derivanti dalla potenziale eliminazione delle trasfusioni per le persone affette da beta-talassemia trasfusione-dipendente e delle crisi vaso-occlusive per quelle affette da anemia falciforme”, dichiara Franco Locatelli, professore di Pediatria presso l’Università Cattolica del Sacro Cuore di Roma, Direttore del Dipartimento di Ematologia e Oncologia Pediatrica dell’Ospedale Pediatrico Bambino Gesù.
Exa-cel è una terapia cellulare non virale, con modifica genica ex vivo mediante tecnologia CRISPR/Cas9, destinata a pazienti elegibili con TDT o SCD, in cui le cellule staminali e progenitrici ematopoietiche del paziente stesso vengono modificate nella regione enhancer specifica per gli eritroidi del gene BCL11A attraverso una precisa rottura del doppio filamento di DNA. Questa modifica porta alla produzione di alti livelli di emoglobina fetale (HbF; emoglobina F) nei globuli rossi. L’HbF è la forma di emoglobina che trasporta l’ossigeno, naturalmente presente durante lo sviluppo fetale, che poi viene sostituita dalla forma adulta di emoglobina dopo la nascita. È stato dimostrato che exa-cel riduce o elimina le VOC nei pazienti con SCD e allevia la necessità di trasfusioni nei pazienti con TDT. Exa-cel è approvato da diversi Enti regolatori in tutto il mondo per i pazienti idonei con SCD e TDT di età pari o superiore a 12 anni.
Grazie a oltre cinque anni di follow-up nei pazienti trattati, gli studi CLIMB-111 e CLIMB-121 hanno mostrato risultati straordinari:
• Il 93% dei pazienti con SCD è risultato libero da crisi vaso-occlusive (VOC) per almeno un anno.
• Il 98% dei pazienti con TDT ha raggiunto l’indipendenza dalle trasfusioni per almeno 12 mesi consecutivi.
Ad oggi, exa-cel è approvato in diverse nazioni, tra cui Stati Uniti, Unione Europea, Canada e Arabia Saudita, con piani per estendere la disponibilità ad altri Paesi. Più di 45 centri di trattamento sono stati attivati, mentre accordi innovativi come quello stipulato con i Centers for Medicare & Medicaid Services (CMS) negli Stati Uniti garantiscono un accesso più ampio e sostenibile per i pazienti.
Talassemia Trasfusione dipendente (TDT): la ferritina può essere correlata alla funzione polmonare
Secondo i risultati di uno studio pubblicati sulla rivista Heliyon , livelli elevati di ferritina sierica sembrano essere correlati alla presenza di disfunzione polmonare nei pazienti giovani con talassemia dipendente da trasfusione (TDT). I pazienti nell'analisi erano candidati al trapianto di cellule staminali emopoietiche ed erano stati sottoposti a test di funzionalità polmonare, esami del sangue e altre valutazioni cliniche.
Sono stati inclusi nell'analisi 140 pazienti con TDT, tra cui 53 (37,9%) pazienti erano donne e la popolazione totale dei pazienti aveva un'età media di 8,7 anni (DS, 3,2 anni). Il livello mediano di ferritina sierica era di 3791,4 ng/mL (intervallo interquartile [IQR], 2424,1-5733,3 ng/mL) e il livello di ferritina sierica era superiore a 2500 ng/mL nel 73,6% dei pazienti. Il livello mediano di emoglobina era di 99,0 g/L (IQR, 90,0-112,5 g/L). Il deposito di ferro epatico è stato identificato in circa l'86% dei pazienti e in grado grave nel 41,4% dei pazienti, con deposito di ferro cardiaco riportato nel 16,4% dei pazienti.
Sono state osservate anomalie polmonari in 65 (46,43%) pazienti. Queste consistevano più comunemente in disfunzione di diffusione (nel 26,43% dei pazienti), deficit misto della funzionalità polmonare (nel 15,0%) e disfunzione ventilatoria (nel 5,0%). La solida correlazione osservata tra livelli elevati di ferritina sierica e disfunzione polmonare nei pazienti con TDT sottolinea l'imperativo di incorporare sistematicamente valutazioni complete della funzionalità polmonare nel monitoraggio clinico di routine di questi pazienti", hanno scritto i ricercatori dello studio nel loro rapporto. Tra i fattori valutati, la funzionalità polmonare anormale è stata associata a un'età più avanzata, a una conta dei globuli rossi più bassa, a un livello di emoglobina più basso, a un deposito di ferro più grave nel cuore e nel fegato e a una maggiore percentuale di pazienti con ferritina sierica superiore a 2500 ng/mL. Dall'analisi si rileva che avere un livello di ferritina sierica superiore a 2500 ng/mL ha mostrato un'associazione significativa con la funzionalità polmonare anomala.
Il farmaco MITAPIVAT

Agios Pharmaceuticals ha annunciato pochi giorni fa il completamento con successo dell'arruolamento dei #pazienti nel suo studio clinico di fase 3 RISE UP, che valuta l'efficacia e la sicurezza del farmaco orale sperimentale #mitapivat nel trattamento della #anemia falciforme in soggetti di età pari o superiore a 16 anni. Lo studio di fase 3 RISE UP ha arruolato oltre 200 pazienti in totale. Gli endpoint primari dello studio includono la risposta dell'emoglobina e il tasso annualizzato di crisi dolorose da anemia falciforme, che sono indicatori significativi dell'impatto della malattia sulla qualità della vita del paziente.
L'azienda prevede di comunicare i risultati principali di questo studio completo di 52 settimane alla fine del 2025. Il farmaco mitapivat ha mostrato risultati promettenti in due studi di fase III per la talassemia trasfusione-dipendente (TDT) e la talassemia non trasfusione-dipendente (NTDT), pertanto Agios Pharmaceuticals sta pianificando di presentare una domanda di commercializzazione alle autorità di regolamentazione negli USA e nell'UE entro la fine del 2024, per il mitapivat destinato ai pazienti con talassemia alfa o beta.
Agios è il leader pioniere nell'attivazione PK e si dedica allo sviluppo e alla fornitura di terapie trasformative per i pazienti affetti da malattie rare. Negli Stati Uniti, Agios commercializza un attivatore della piruvato chinasi (PK) first-in-class per adulti con deficit di PK, la prima terapia modificatrice della malattia per questa rara, debilitante e permanente anemia emolitica. Basandosi sulla profonda competenza scientifica dell'azienda in ematologia classica e sulla leadership nel campo del metabolismo cellulare e delle malattie ematologiche rare, Agios sta promuovendo una solida pipeline clinica di medicinali sperimentali con programmi in alfa e beta-talassemia, anemia falciforme, deficit di PK pediatrico, anemia associata alla sindrome mielodisplastica (MDS) e fenilchetonuria (PKU). Oltre alla sua pipeline clinica, Agios sta promuovendo un siRNA TMPRSS6 preclinico come potenziale trattamento per la policitemia vera. Per maggiori informazioni www.agios.com
Novartis cede i diritti mondiali sul Desferal (deferoxamina)

Mitem Pharma ha acquistato da Novartis i diritti mondiali sul Desferal (deferoxamina), un trattamento utilizzato soprattutto come chelante per abbassare livelli eccessivi e dannosi di ferro indotti dalle trasfusioni di sangue nei soggetti affetti da anemia falciforme o nella beta #talassemia e altre anemie. L'acquisizione è stata raggiunta in un accordo tra Mitem e Novartis, che ha sviluppato e inizialmente commercializzato il farmaco #Desferal.
Le trasfusioni di sangue di routine possono portare a complicazioni come il sovraccarico di ferro (emocromatosi), con un accumulo eccessivo di questo minerale essenziale, che può danneggiare organi come il cuore e il fegato. Desferal è una terapia chelante del ferro somministrata tramite iniezione sottocutanea (sotto la pelle).
Mitem, con sede in #Francia, è specializzata nell'identificazione e nel mantenimento delle scorte di medicinali essenziali prescritti per malattie gravi, i cosiddetti medicinali di maggiore interesse terapeutico le cui difficoltà di approvvigionamento comporterebbero un rischio significativo per i pazienti e un rischio per la salute pubblica. Presente in 15 paesi di 3 continenti, MITEM PHARMA è un laboratorio farmaceutico di nuova generazione la cui missione è migliorare la qualità della vita dei pazienti garantendo la fornitura di farmaci importanti, gli unici trattamenti per malattie gravi, ma la cui carenza di offerta causa rischi per i pazienti. DESFERAL® (deferoxamina), è un medicinale iniettabile incluso nell'elenco dei farmaci essenziali dell'Organizzazione Mondiale della Sanità e commercializzato in più di 60 paesi, è indicato in caso di sovraccarico di ioni metallici, in particolare a seguito di trasfusioni di sangue.
La scoperta di un nuovo gruppo sanguigno
Il team di ricerca, guidato dagli scienziati dell'NHS Blood and Transplant (NHSBT) nel South Gloucestershire e supportato dall'Università di Bristol, ha scoperto un gruppo sanguigno chiamato MAL. Hanno identificato il background genetico dell'antigene del gruppo sanguigno AnWj, precedentemente noto, scoperto nel 1972 ma sconosciuto fino ad ora dopo lo sviluppo di questo primo test al mondo. Migliaia di vite potrebbero essere salvate in tutto il mondo dopo che i ricercatori hanno scoperto un nuovo sistema di gruppi sanguigni, risolvendo un mistero vecchio di 50 anni.
Tutti noi abbiamo delle proteine esterne ai globuli rossi chiamate antigeni, ma in alcuni casi potrebbero esserne privi. Utilizzando test genetici, il Laboratorio di riferimento internazionale per i gruppi sanguigni dell'NHSBT di Filton ha sviluppato per la prima volta un test in grado di identificare i pazienti privi di questo antigene. Il test potrebbe rivelarsi una salvezza per coloro che avrebbero una reazione avversa a una trasfusione di sangue e renderà più facile trovare potenziali sviluppatori di sangue per questo raro gruppo sanguigno.
Il team di ricerca ha affermato "Tutto ciò che possiamo fare per rendere il nostro sangue più sicuro e più adatto a quello dei pazienti è sicuramente un passo nella giusta direzione. Ora è possibile progettare test di genotipizzazione per identificare pazienti e donatori geneticamente AnWj-negativi. Tali test possono essere aggiunti alle piattaforme di genotipizzazione esistenti.
Fonte https://www.bbc.com/news/articles/
Aifa ritira un lotto di Oxbryta, medicinale indicato per l’anemia falciforme
L’Aifa, con provvedimento del 30.9.2024, ha disposto il ritiro del medicinale OXBRYTA 500 mg 90 compresse, AIC. 049971017, lotto n. 2026284 scad. 30 aprile 2026 della ditta Pfizer Srl.
Il provvedimento è stato disposto, a scopo cautelativo, a seguito della comunicazione da parte della ditta Pfizer srl in cui informa della raccomandazione del Comitato per i medicinali per uso umano dell’Ema di voler sospendere l’autorizzazione all’immissione in commercio del medicinale Oxbryta (voxelotor) a titolo precauzionale per la durata della revisione dei dati emergenti relativi alla maggiore incidenza di crisi vaso-occlusive (VOC) durante il trattamento con Oxbryta rispetto a prima della terapia.
Oxbryta è utilizzato per il trattamento dell’anemia emolitica (ossia eccessiva degradazione dei globuli rossi) dovuta ad anemia falciforme in pazienti a partire dai 12 anni di età. Oxbryta può essere somministrato da solo o insieme a un altro medicinale per l’anemia falciforme chiamato idrossicarbamide.
Approvato negli Stati Uniti con un processo accelerato nel 2019 e successivamente nell'UE nel febbraio 2022 , il farmaco inizialmente si è mostrato promettente nell'alleviare le dolorose complicazioni associate alla SCD. Oxbryta, progettato per trattare l'anemia falciforme, è stato un farmaco cruciale per i pazienti con sintomi da lievi a moderati.
Tuttavia, recenti dati clinici hanno messo in dubbio la sua sicurezza ed efficacia complessive. La sospensione di Oxbryta segna un momento significativo nella battaglia in corso contro l'anemia falciforme. Mentre la comunità medica continua a cercare trattamenti efficaci per questa condizione debilitante, la sicurezza del paziente rimane fondamentale.
La richiesta dell'EMA di sospendere l'uso del farmaco è stata influenzata dai dati di sicurezza emergenti da due studi basati su registri. Questi studi hanno rivelato che i pazienti hanno sperimentato una maggiore incidenza di VOC durante il trattamento con Oxbryta rispetto a prima di iniziare il farmaco. Data la gravità di questi risultati, l'EMA ha concluso che i rischi associati a Oxbryta ora superano i suoi benefici.
L'editing genetico: la storia di Michele

Roma – Michele ha una storia che pochi altri al mondo possono raccontare: una storia di scienza, medicina e innovazione, che ha visto come protagonista un incontro fortuito tra le sue cellule staminali ematopoietiche e CRISPR. Michele è, infatti, uno dei giovanissimi pazienti con Ɓtalassemia che ha partecipato allo studio clinico internazionale CLIMB-111, realizzato per valutare la terapia basata su CRISPR-Cas9 e che in Italia ha coinvolto l’Ospedale Pediatrico Bambino Gesù di Roma. Michele non ha fatto trasfusioni per più di un anno, ma poi ha dovuto sottoporsi alla procedura a settembre 2023, a causa di una brutta gastroenterite. Dal 17 settembre 2023 è completamente libero dalle trasfusioni.
L’indipendenza dalle trasfusioni periodiche, che sono necessarie per mantenere i valori di emoglobina in un range di non pericolosità per l’organismo, è il grande impatto positivo di questa terapia: le forbici molecolari più famose al mondo, infatti, hanno permesso al 91% dei pazienti con beta-talassemia coinvolti nello studio di non dover più sottoporsi alle trasfusioni, guadagnando così tempo e qualità di vita.
L’editing genomico è una innovativa e versatile tecnologia utilizzata in tutti i laboratori del mondo per modificare il DNA ed è diventata una rivoluzionaria terapia per chi ha una diagnosi di beta-talassemia o di anemia falciforme. Un trattamento che permette di intervenire sul gene BCL11A, che regola la produzione di emoglobina nel sangue al termine della vita fetale. Nel feto, di fatto, è presente una forma di emoglobina diversa, che viene progressivamente sostituita dalla nascita proprio grazie all’azione del gene BCL11A. Exagamglogene autotemcel, il sistema CRISPR, si basa sul ripristino della sintesi dell’emoglobina fetale, andando a spegnere il gene BCL11A.
"La malattia toglie tanto, ma insegna anche tanto. Quando mio figlio ha ricevuto la diagnosi già si parlava di una terapia, ma ancora non c’era nulla di concreto per i pazienti e i tempi non si potevano prevedere. Vorrei anche ringraziare la dott.ssa Iacono, Presidente della Fondazione Italiana Talassemia e Drepanocitosi “Leonardo Giambrone” (FITHAD): tutto questo non sarebbe stato possibile senza di lei”, conclude la mamma Maria. “Noi genitori non riusciremo mai a dimenticare quello che abbiamo passato fin dai primi mesi di vita di Michele, ma lui sì. L’ha già dimenticato e ha tutta la vita davanti”.
Leggi la storia su
La Commissione Europea autorizza Casgevy una terapia di editing genico

13 febbraio 2024 - Vertex Pharmaceuticals Incorporated ha annunciato che la Commissione Europea ha concesso l’autorizzazione all’immissione in commercio condizionata per Casgevy (exagamglogene autotemcel -exa-cel), una terapia di editing genico basata su tecnologia Crispr/Cas9. Casgevy è approvato per il trattamento di pazienti di età pari o superiore a 12 anni affetti da beta-talassemia dipendente dalle trasfusioni (Tdt) oppure da anemia falciforme severa (Scd) caratterizzata da crisi vaso-occlusive (Voc) ricorrenti, per i quali è appropriato il trapianto di cellule staminali ematopoietiche (Hsc) e non è disponibile un donatore consanguineo con antigene leucocitario umano (Hla) compatibile.
Casgevy è attualmente l’unica terapia genica approvata per i pazienti affetti da SCD e TDT nell’UnioneEuropea e con questa approvazione saranno ora oltre 8.000 i pazienti potenzialmente eleggibili altrattamento. «Grazie a questa approvazione, Casgevy è ora disponibile per l’anemia falciforme e la beta-talassemia dipendente dalle trasfusioni in diverse aree geografiche, rendendo decine di migliaia di pazienti eleggibili per questa terapia in grado di offrire un’alternativa terapeutica efficace per cambiare il corso naturale delle due patologie - ha dichiarato Reshma Kewalramani, Ceo e Presidente di Vertex -.Ora il nostro obiettivo è tradurre queste approvazioni in benefici tangibili per i pazienti, garantendo l’accesso e la rimborsabilità in tutto il mondo». «L’anemia falciforme e la beta-talassemia dipendente dalle trasfusioni sono patologie ereditarie del globulo rosso, che hanno un impatto significativo sull’aspettativa e sulla qualità di vita dei pazienti e delle loro famiglie, con importanti ricadute anche sui sistemi sanitari - ha dichiarato Franco Locatelli, sperimentatore principale degli studi Climb-111 e Climb-121, professore di Pediatria presso l’Università Cattolica del Sacro Cuore di Roma e Direttore del Dipartimento di Ematologia e Oncologia Pediatrica dell’Ospedale Pediatrico Bambino Gesù -. Casgevy ha il potenziale per trasformare la vita dei pazienti affetti da queste due patologie; ora è importante che questa terapia sia messa rapidamente a disposizione dei pazienti eleggibili».
Vertex sta già collaborando con le autorità regolatorie nazionali per garantire un accesso rapido ai pazienti eleggibili. In Francia questa collaborazione ha portato all’accesso precoce (early access) di Casgevy per il trattamento della Ttd. Ciò significa che tutti i pazienti eleggibili in Francia potranno accedere gratuitamente alla terapia prima del suo rimborso a carico del Servizio sanitario nazionale francese. Vertex sta inoltre collaborando con ospedali specializzati nei trapianti di cellule staminali con l’obiettivo di creare una rete di centri di trattamento autorizzati (Atc), pubblici e privati, per la somministrazione di Casgevy. Attualmente sono tre gli Atc attivati nell’Ue. L’azienda prevede di attivarne un totale di circa 25 in tutta Europa. Casgevy è una terapia cellulare non virale, con modifica genica ex vivo mediante tecnologia Crispr/Cas9,destinata a pazienti eleggibili con Tdt o Scd, in cui le cellule staminali e progenitrici ematopoietiche del paziente stesso vengono modificate nella regione enhancer specifica per gli eritroidi del gene Bcl11A attraverso una precisa rottura del doppio filamento di Dna. Questa modifica porta alla produzione di alti livelli di emoglobina fetale (HbF; emoglobina F) nei globuli rossi. L’HbF è la forma di emoglobina che trasporta l’ossigeno, naturalmente presente durante lo sviluppo fetale, che poi viene sostituita dalla forma adulta di emoglobina dopo la nascita. È stato dimostrato che Casgevy riduce o elimina le Voc nei pazienti con Scd e allevia la necessità di trasfusioni nei pazienti con Tdt.
